
Health May 29, 2026
Bioequivalence Studies: What FDA Requires Manufacturers to Prove
When you pick up a prescription at the pharmacy, there is a very high chance it comes in plain white packaging with no flashy brand logo. That is a generic drug. But how do we know it works exactly like the expensive brand-name version? The answer lies in bioequivalence studies, which are clinical trials designed to prove that a generic drug delivers the same amount of active ingredient into your bloodstream as the original brand-name drug. Without these rigorous tests, the entire system of affordable healthcare would crumble. The U.S. Food and Drug Administration (FDA) does not just take manufacturers' word for it. They demand hard data.
If you are a pharmaceutical manufacturer looking to launch a generic product, understanding what the FDA requires is not optional-it is the difference between a successful launch and a rejected application. This guide breaks down exactly what you need to prove, how the testing works, and where most companies make costly mistakes.
The Core Requirement: Proving Therapeutic Equivalence
At its heart, the goal of a bioequivalence study is simple: show that the generic drug behaves in the body just like the Reference Listed Drug (RLD), which is the original brand-name drug approved by the FDA that serves as the standard for comparison. Under the Hatch-Waxman Amendments of 1984, manufacturers can skip repeating massive clinical safety trials if they prove their product is bioequivalent to an already-approved RLD.
To meet this requirement, two things must be true:
- Pharmaceutical Equivalence: Your drug must have the same active ingredient, dosage form (like a tablet or capsule), strength, and route of administration as the RLD.
- Bioequivalence: The rate and extent to which the active ingredient becomes available in your body must be statistically similar to the RLD.
The FDA defines bioequivalence specifically as the absence of a significant difference in the rate and extent of absorption when administered at the same molar dose under similar conditions. This definition is codified in Title 21 of the Code of Federal Regulations (21 CFR 320.1). It is not enough for the pill to look the same; it must perform the same.
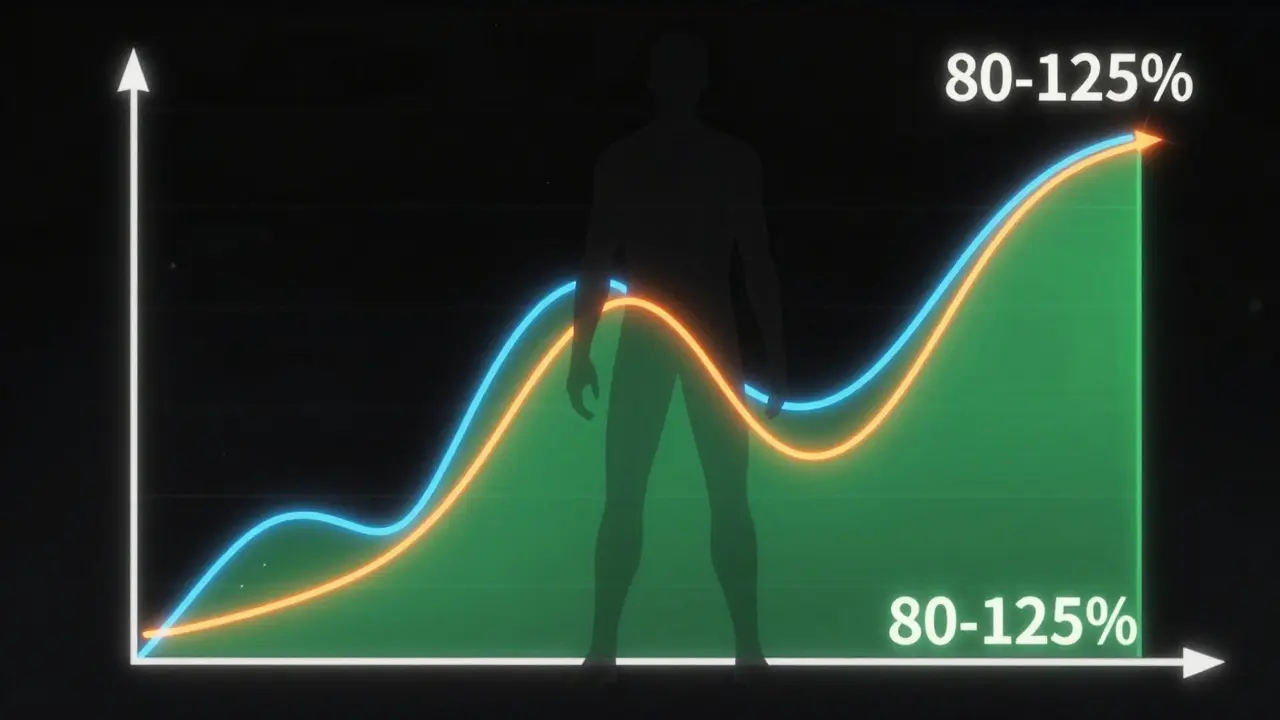
The 80/125 Rule: The Golden Standard of Approval
You cannot eyeball bioequivalence. You need statistics. The FDA uses pharmacokinetic endpoints-specifically, how much drug gets into the blood and how fast-to judge success. The two main metrics are:
- AUC (Area Under the Curve): This measures the total exposure to the drug over time. It tells us how much of the drug actually got into the system.
- Cmax (Maximum Plasma Concentration): This measures the peak level of the drug in the blood. It tells us how quickly the drug was absorbed.
Here is the critical part: the FDA requires that the 90% confidence interval for the ratio of the geometric means of these metrics (generic vs. brand) falls within a specific range. That range is 80% to 125%. This is known as the "80/125 rule."
Why those numbers? Because after log-transformation, this range represents a symmetric window around 100%. If your generic drug’s AUC and Cmax fall within this bracket compared to the brand, the FDA considers them therapeutically equivalent. This rule has been the cornerstone of generic drug approval since 1992. For most systemic drugs, if you miss this window, your application fails.
| Metric | What It Measures | FDA Acceptance Criteria |
|---|---|---|
| AUC | Total drug exposure over time | 90% CI within 80-125% |
| Cmax | Peak concentration in blood | 90% CI within 80-125% |
| Tmax | Time to reach peak concentration | Descriptive only (not used for statistical pass/fail) |
Study Design: How Many Volunteers Do You Need?
Most bioequivalence studies are conducted in healthy volunteers, not patients. The typical sample size ranges from 24 to 36 subjects. These studies usually follow a crossover design, meaning each volunteer receives both the generic and the brand-name drug in separate periods, with a washout period in between to ensure the first drug leaves their system completely.
The studies must be conducted under fasting conditions unless the drug is known to interact significantly with food. In such cases, the FDA may require additional fed-state studies. Every step of this process-from sample collection to storage-must adhere to Good Laboratory Practice (GLP) regulations outlined in 21 CFR Part 58. Poor documentation here is one of the top reasons for rejection.
Exceptions: When You Can Skip Clinical Trials (Biowaivers)
Not every generic drug needs a full-blown human trial. The FDA allows biowaivers, which are regulatory exemptions that allow manufacturers to waive in vivo bioequivalence studies for certain types of products. This saves time and money, but the criteria are strict.
According to 21 CFR 320.22, you might qualify for a biowaiver if your product meets the Q1-Q2-Q3 framework:
- Q1: Identical active and inactive ingredients as the RLD.
- Q2: Same dosage form and concentration.
- Q3: Equivalent pH and physicochemical properties.
This often applies to parenteral solutions, ophthalmic drops, or topical products intended for local effect rather than systemic absorption. For complex topicals, the FDA may accept in vitro release testing (IVRT) instead of human trials. As of late 2023, the FDA had issued over 1,200 product-specific guidances detailing which products qualify. Always check the latest guidance before designing your study.
Special Cases: Narrow Therapeutic Index Drugs
The 80/125 rule works well for most drugs, but some medications are unforgiving. These are called Narrow Therapeutic Index Drugs (NTIDs), which are medications where small differences in blood levels can lead to treatment failure or serious toxicity. Examples include warfarin (a blood thinner) and levothyroxine (for thyroid conditions).
For NTIDs, the FDA tightens the acceptance criteria. Instead of 80-125%, the 90% confidence interval must fall within 90% to 111%. This narrower window ensures patient safety for drugs where precision is critical. If you are developing a generic for an NTID, your statistical power must be higher, and your manufacturing controls must be exceptionally tight.

Common Pitfalls That Lead to Rejection
Even with a solid plan, many applications fail. In fiscal year 2022, the FDA’s first-cycle approval rate for Abbreviated New Drug Applications (ANDAs) was only 43%. Here is why:
- Inadequate Study Design: Choosing the wrong number of volunteers or failing to account for high variability in drug absorption.
- Poor Analytical Methodology: Using assays that are not sensitive enough to detect low concentrations of the drug accurately.
- Ignoring Product-Specific Guidance (PSG): The FDA has published thousands of PSGs. Companies that follow these guidelines see a 68% first-cycle approval rate, compared to just 29% for those who don’t.
- Documentation Gaps: Missing records on sample handling or deviations from the protocol during the trial.
Cost is also a factor. A single bioequivalence study can cost between $500,000 and $2 million. Wasting that budget on a flawed design is devastating. Invest in expert biostatisticians early in the process.
The Future: Complex Generics and New Tools
The landscape is shifting. With the Generic Drug User Fee Amendments (GDUFA III) running through 2027, the FDA is focusing heavily on complex generics like inhalers, patches, and drug-device combinations. Traditional PK studies often don't work for these products.
The FDA is increasingly accepting mechanistic tools like physiologically based pharmacokinetic (PBPK) modeling to support bioequivalence assessments. This computational approach simulates how drugs move through the body, offering a faster alternative to some clinical trials. Additionally, the Domestic Generic Drug Manufacturing Pilot Program offers expedited review for generics made with U.S.-sourced ingredients and tested domestically, potentially cutting the typical 10-month review timeline.
Final Checklist for Manufacturers
Before you submit your ANDA, run through this quick checklist:
- Have you identified the correct Reference Listed Drug (RLD)?
- Does your study design align with the latest Product-Specific Guidance (PSG)?
- Are your statistical methods powered to meet the 80/125 (or 90/111 for NTIDs) criteria?
- Is your GLP documentation complete and audit-ready?
- Have you considered if a biowaiver applies to save time and resources?
Bioequivalence is not just a regulatory hurdle; it is the scientific proof that keeps healthcare accessible. By following the FDA’s strict requirements, you ensure that patients get safe, effective, and affordable treatments.
What is the primary purpose of a bioequivalence study?
The primary purpose is to demonstrate that a generic drug performs comparably to the reference listed drug (brand-name) in terms of the rate and extent of absorption. This ensures therapeutic equivalence without requiring new clinical safety trials.
What does the 80/125 rule mean in bioequivalence?
The 80/125 rule states that the 90% confidence interval for the ratio of geometric means of key pharmacokinetic parameters (AUC and Cmax) between the generic and brand drug must fall between 80% and 125%. This indicates no significant difference in performance.
Can I skip clinical trials for my generic drug?
Yes, if your product qualifies for a biowaiver. This typically applies to simple formulations like solutions or topicals with identical ingredients and properties to the reference drug. You must meet strict Q1-Q2-Q3 criteria.
How many volunteers are needed for a standard bioequivalence study?
Most standard studies require between 24 and 36 healthy volunteers. The exact number depends on the variability of the drug and the statistical power required to meet FDA acceptance criteria.
What are Narrow Therapeutic Index Drugs (NTIDs)?
NTIDs are drugs where small changes in blood concentration can cause toxicity or treatment failure. Examples include warfarin and levothyroxine. The FDA requires tighter bioequivalence limits (90-111%) for these drugs.
Write a comment
Items marked with * are required.



12 Comments
Anthony Padilla May 31, 2026 AT 06:13
Hey guys, just wanted to share some thoughts on this because it really hits home for me. I always wondered how they make sure the cheap pills are safe. It is crazy to think about all the testing that goes into something so small. The part about the 80/125 rule was super interesting to read through. It makes you realize that there is a lot of science behind what we swallow every day. We should be more grateful for the people who do these studies honestly. It keeps healthcare affordable for everyone which is huge. I hope this helps clarify things for anyone else confused about generics.
Elizabeth Fandry May 31, 2026 AT 15:25
One must ponder the philosophical implications of therapeutic equivalence in the modern age 🧐💊 Is it truly equivalent if the soul of the brand is absent? The FDA’s rigid adherence to statistical boundaries suggests a mechanistic view of human biology that ignores the subtle nuances of individual constitution. To reduce life-saving medication to mere AUC and Cmax metrics is a profound reductionism. We are not merely vessels for chemical absorption; we are complex beings deserving of holistic consideration. Yet here we are, bound by the 80/125 rule as if it were divine law. 😒📉
Madeline Petes June 1, 2026 AT 22:13
This is such an awesome breakdown!! I never knew about the biowaivers before reading this. It's so cool that you can skip trials for some stuff like eye drops or topicals. Makes total sense why they don't need full human trials for those. I feel way smarter now lol. Thanks for sharing this info it really helps understand the process better. You did a great job explaining it simply without being boring at all!
Ramanath Rao June 2, 2026 AT 17:27
In India we have similar processes but the enforcement is sometimes lacking compared to US standards. This article highlights the rigor required which is good to see. However many manufacturers still cut corners globally. The 80/125 rule is standard but implementation varies wildly across different regulatory bodies. We need stricter global harmonization to ensure patient safety everywhere not just in wealthy nations. The cost mentioned here is high but necessary for quality control.
irine sabrina June 3, 2026 AT 12:55
I am so glad someone wrote this because it takes away a lot of fear about taking generic meds. Knowing that there are strict rules gives me peace of mind when picking up prescriptions. It is reassuring to know that experts are watching over the process closely. Let us keep supporting affordable healthcare options for our communities. Everyone deserves access to safe medications regardless of their income level. Thank you for spreading awareness on this important topic today.
Gary Helminiak June 5, 2026 AT 04:24
As someone who works in pharma QA, I can tell you that the documentation gaps mentioned are absolutely real and frustrating. We spend weeks just ensuring GLP compliance because one missing signature can tank a whole study. The PBPK modeling part is the future though. It saves so much time and resources compared to traditional crossover designs. Plus fewer volunteers means less ethical complexity. Just make sure your statistical power is calculated correctly upfront or you will end up running extra subjects which costs way more money in the long run. Trust me on this one. 👍📊
dane thorp June 5, 2026 AT 14:28
The section on Narrow Therapeutic Index Drugs is particularly critical. Warfarin and levothyroxine require precision that leaves no room for error. The tighter 90-111% window reflects the clinical reality where small deviations matter significantly. Manufacturers must respect these constraints strictly.
Michael Schurmann June 7, 2026 AT 03:10
It is amusing how laypeople assume generics are identical to brands. They are statistically similar within a wide margin. If you care about health outcomes you pay for the original. The 80/125 rule allows for significant variation that most patients ignore. Big pharma knows this which is why they lobby against biosimilars too. Wake up sheeple.
Christina Mitchell June 7, 2026 AT 09:50
We live in a world where trust is currency. These studies build that trust between patients and providers. It is beautiful to see science serving humanity in such a practical way. May we continue to value evidence-based medicine above all else. Peace and love to all seeking knowledge.
Christopher Laver June 7, 2026 AT 09:56
Boring. Read the first paragraph and skipped the rest. Too much jargon for my taste. Just tell me if it works or not.
Russell Russell June 8, 2026 AT 05:11
Let us break down the importance of Product-Specific Guidance again. Ignoring PSG is the number one reason for failure. Do not be lazy with your research. Check the FDA database daily. Success comes from preparation and attention to detail. Keep pushing forward and never settle for mediocrity in your applications.
Naresh Chandra June 9, 2026 AT 03:04
I appreciate the detailed explanation regarding the Q1-Q2-Q3 framework! It clarifies the criteria for biowaivers significantly. Many professionals overlook these specific requirements leading to unnecessary delays. Thank you for highlighting this crucial aspect of regulatory strategy. It is indeed vital for efficient development pipelines.